GWAS with nf-gwas
nf-gwas is a Nextflow pipeline designed for biobank-scale genome-wide association studies (GWAS). The pipeline automatically performs multiple pre- and post-processing steps, integrates regression modeling from the REGENIE package, and currently supports single-variant, gene-based, and interaction testing. All modules are structured as sub-workflows, allowing future extensions to other methods and tools. nf-gwas includes extensive reporting functionality, enabling users to inspect thousands of phenotypes and navigate interactive Manhattan plots directly in a web browser.
Prerequisites
The following software is required to set up your local imputation workflow. This has been tested on various Linux distributions as well as macOS:
- Java 17 or higher
- Nextflow
- Docker or Singularity
Setup
First, we need to prepare the input files for nf-gwas. The files are also available here.
mkdir nf-gwas-test
cd nf-gwas-test
# Download phenotype and covariates
wget https://genepi.i-med.ac.at/downloads/imputation/phenotypes.txt
wget https://genepi.i-med.ac.at/downloads/imputation/covariates.txt
# Download imputed data
wget https://genepi.i-med.ac.at/downloads/imputation/gwas.imputed.chr20.dose.vcf.gz
# Download annotation files for chr20
wget https://genepi.i-med.ac.at/downloads/imputation/rsids-v154-hg19-chr20.index.gz .
wget https://genepi.i-med.ac.at/downloads/imputation/rsids-v154-hg19-chr20.index.gz.tbi .nf-gwas Parameters
The nf-gwas pipeline supports a variety of configurable parameters.
In the examples below, we demonstrate a simplified GWAS analysis where we skip Step 1 of REGENIE — the model-fitting stage.
Step 1 of REGENIE usually fits a ridge regression model to predict the phenotype (or residuals) from genotypes, accounting for population structure and relatedness. Since regenie_skip_predictions = true, this step is bypassed, and we proceed directly to Step 2, which performs the association testing.
Below is a breakdown of the main parameters:
| Parameter | Description |
|---|---|
project |
A name for the analysis run. Useful for organizing output files. |
genotypes_association |
The genotype file used in Step 2. Here, it’s a compressed VCF file (.vcf.gz) containing imputed genotypes for chromosome 20. |
genotypes_association_format |
Specifies the format of the genotype file (vcf here). Internally, nf-gwas converts this VCF into PLINK2 format for REGENIE. |
association_build |
The genome reference build used (hg19 or hg38). |
phenotypes_filename |
File containing phenotype data for each individual. |
phenotypes_columns |
Specific phenotype columns to include (pheno_2 and pheno_4). |
phenotypes_binary_trait |
Whether phenotypes are binary (true) or continuous (false). |
covariates_filename |
File listing covariates (e.g., PCs) to adjust for population stratification. |
covariates_columns |
Covariate columns to include (PC1–PC10). |
regenie_test |
Genetic model to test; here, additive. |
regenie_min_imputation_score |
Minimum genotype imputation quality score; variants below are excluded. |
rsids_filename |
Index file mapping variant positions to rsIDs for annotation. |
binning_size |
Genomic bin size (in base pairs) for plotting and indexing results. |
This configuration performs a straightforward GWAS on two continuous traits (pheno_2 and pheno_4), adjusting for population structure using the top 10 principal components (PCs).
GWAS Without PCA
We first run a linear regression GWAS on a simulated phenotype without adjusting for population structure. Please create a gwas.config file with this content:
params {
project = 'test-gwas'
genotypes_association = 'gwas.imputed.chr20.dose.vcf.gz'
regenie_skip_predictions = true
association_build = 'hg19'
genotypes_association_format = 'vcf'
phenotypes_filename = 'phenotypes.txt'
phenotypes_columns = 'pheno_2,pheno_4'
phenotypes_binary_trait = false
regenie_test = 'additive'
regenie_min_imputation_score = 0.3
rsids_filename = "rsids-v154-hg19-chr20.index.gz"
binning_size = 50000
}Run GWAS
Execute the pipeline:
nextflow run genepi/nf-gwas -r v1.0.11 -profile docker -c gwas.configIf you are running this on a server with Singularity, change the profile to "singularity" instead of "docker".
Results
All results (REGENIE outputs, top hits, graphical reports) are available within the results folder.
GWAS With PCA
Next, we run a linear regression GWAS on a simulated phenotype while adjusting for population structure using principal components. Please create a gwas_with_pcs.config file with this content:
params {
project = 'test-gwas-with-pcs'
genotypes_association = 'gwas.imputed.chr20.dose.vcf.gz'
regenie_skip_predictions = true
association_build = 'hg19'
genotypes_association_format = 'vcf'
phenotypes_filename = 'phenotypes.txt'
phenotypes_columns = 'pheno_2,pheno_4'
phenotypes_binary_trait = false
regenie_test = 'additive'
regenie_min_imputation_score = 0.3
covariates_filename = 'covariates.txt'
covariates_columns = 'PC1,PC2,PC3,PC4,PC5,PC6,PC7,PC8,PC9,PC10'
rsids_filename = "rsids-v154-hg19-chr20.index.gz"
binning_size = 50000
}Run GWAS
Execute the pipeline:
nextflow run genepi/nf-gwas -r v1.0.11 -profile docker -c gwas_with_pcs.configResults
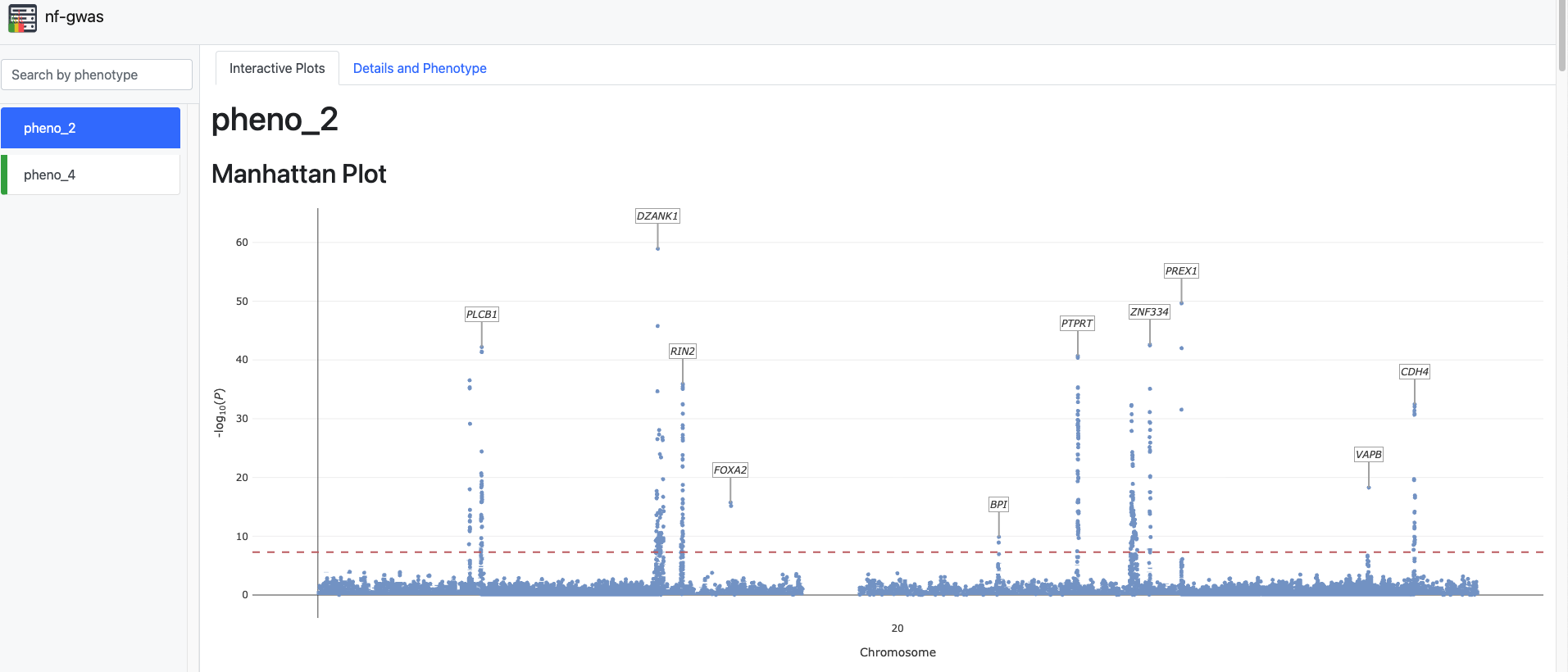
All results (REGENIE outputs, top hits, and graphical reports) are available in the results folder. The folder also includes an HTML file containing an interactive Manhattan plot:
Manhattan Plot without PCs
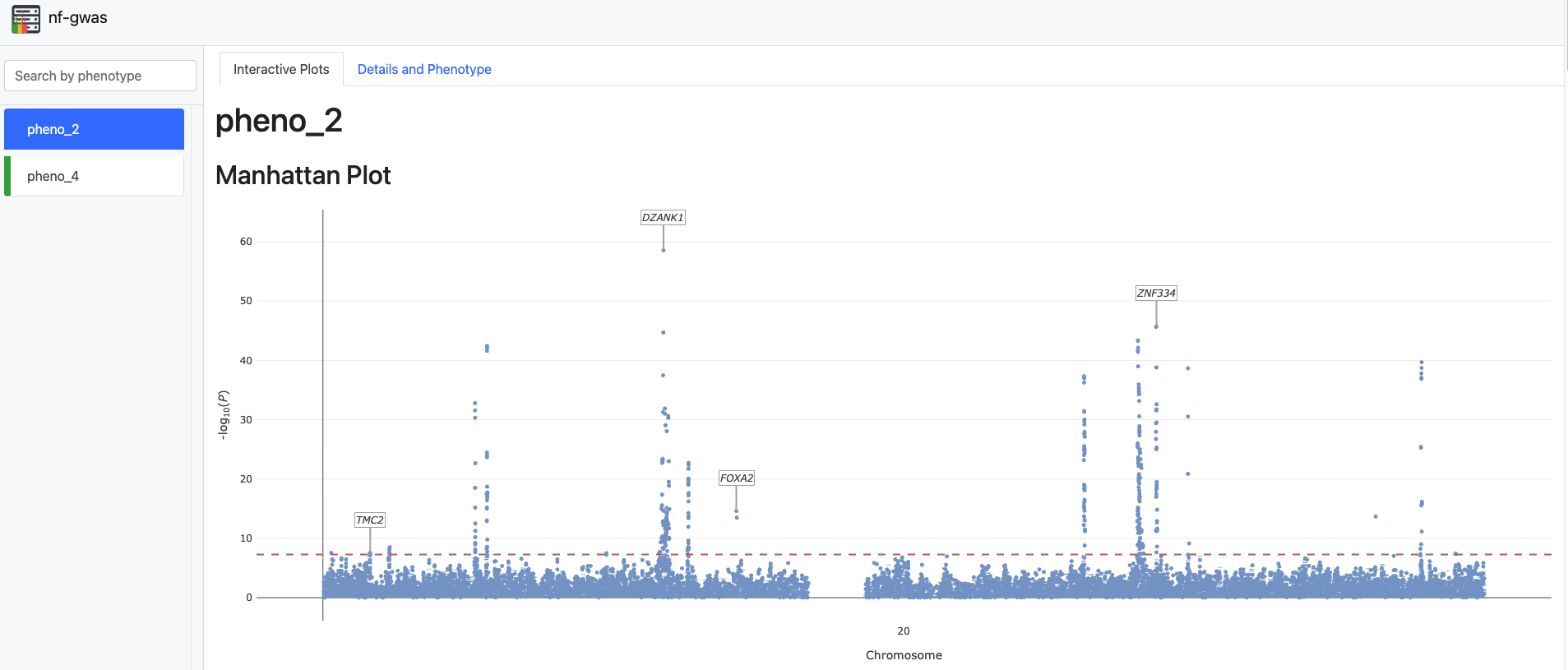
Manhattan Plot with PCs