Welcome to nf-gwas!
A Nextflow pipeline to perform genome-wide association studies (GWAS).
Get started now View it on GitHub
This cloud-ready GWAS pipeline allows you to run single variant tests, gene-based tests and interaction testing using REGENIE in an automated and reproducible way.
For single variant tests, the pipeline works with BGEN (e.g. from UK Biobank) or VCF files (e.g. from Michigan Imputation Server). For gene-based tests, we currently support BED files as an input. The output files of the pipeline include results of the association tests (in tabix indexed format, which works with e.g. LocusZoom out of the box), annotated loci tophits and an interactive HTML report with summary statistics and plots.
The single-variant pipeline currently includes the following steps:
-
Validate phenotype and covariate file (e.g. check file format, replace empty values with NA, create summary statistics)
-
Convert imputed data in VCF format into the plink2 file format (optional).
-
Prune micro-array data using plink2 (optional).
-
Filter micro-array data using plink2 based on MAF, MAC, HWE, genotype missingness and sample missingness.
-
Run regenie and index (tabix) results to use with LocusZoom.
-
Parse regenie log and create summary statistics.
-
Filter regenie results by pvalue.
-
Annotate filtered results using genomic-utils and genes from GENCODE.
-
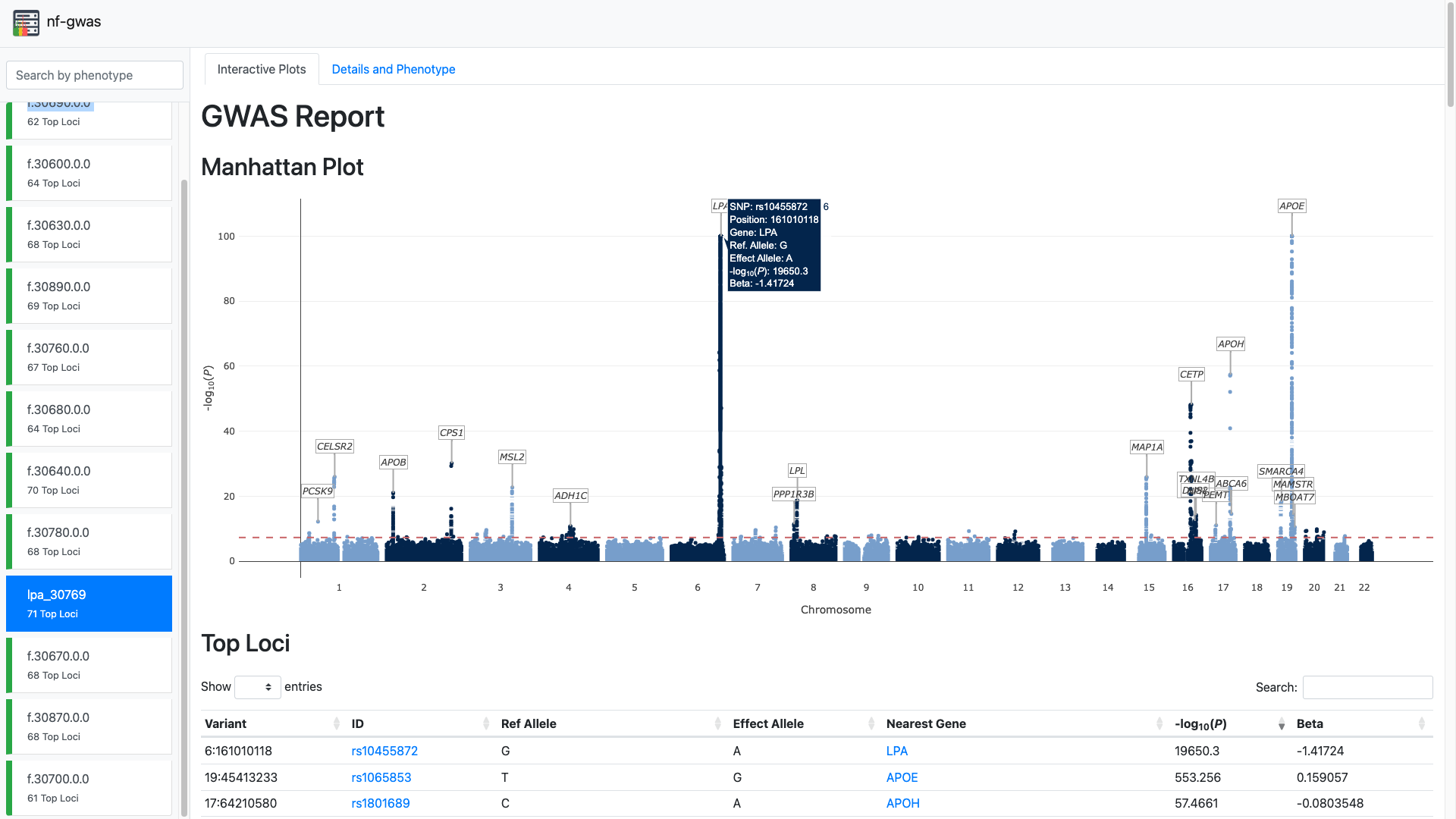
Create a HTML report per phenotype including the annotated manhattan plot, qq plot, top loci, phenotype statistics and parsed log files.